感染組織的宏基因組測序,您選對DNA抽提方法了嗎?
本文轉(zhuǎn)自歐易生物
前言
16S核糖體RNA基因(rRNA)的擴增測序及分析是評價臨床標(biāo)本細菌群落多樣性常用的方法。該方法利用與16S rDNA高度保守區(qū)域結(jié)合的引物,通過PCR擴增16S rRNA基因,對擴增得到的產(chǎn)物進行高通量測序及數(shù)據(jù)分析獲得樣本中細菌群落的OTU,從而判斷樣本中的微生物種類。該方法優(yōu)點是簡便快速,缺點是大部分微生物無法鑒定到種的水平。
隨著測序平臺發(fā)展和測序成本的降低,人們對微生物組成的研究從系統(tǒng)發(fā)育研究(16S rRNA)轉(zhuǎn)向了宏基因組測序。通過覆蓋微生物群落的所有物種的基因組信息,使物種鑒定結(jié)果具有更高的特異性和敏感性。宏基因組測序還可以提供酶組成或代謝途徑、細菌功能基因組成以及功能與系統(tǒng)發(fā)育之間的基因組聯(lián)系的遺傳證據(jù)。另外,宏基因組學(xué)不僅可以分析來自細菌源的序列,還可以分析來自真菌、病毒和寄生蟲的序列。基于上述優(yōu)勢,使得直接分析單個樣本中的微生物組數(shù)據(jù)成為研究臨床樣本微生物多樣性的常用方法。
然而,在研究人類疾病與微生物之間的關(guān)系時,由于宿主DNA的污染,宏基因組測序在實驗上面臨巨大挑戰(zhàn)。宿主DNA與微生物DNA的共提取導(dǎo)致后續(xù)測序數(shù)據(jù)中大部分是宿主的基因組數(shù)據(jù),從而使微生物組的數(shù)據(jù)不足,還可能產(chǎn)生干擾下游分析的非特異性信號。解決這個問題有兩種方法:要么需要去除宿主DNA,要么需要增加測序深度以獲取足夠的微生物基因組數(shù)據(jù)進行后續(xù)分析。然而增加測序深度可能更昂貴,因此我們比較了幾種去除宿主DNA的微生物基因組抽提方法。
材料與方法
1)材料
糖尿病足感染樣本;
2)方法(試劑盒)
實驗設(shè)計及結(jié)果展示
1. 實驗設(shè)計
糖尿病足感染樣本勻漿混樣,平均分成15份,每份25mg,用5種方法抽提DNA,并對所得DNA進行定量PCR及16S rDNA 擴增測序以檢測物種多樣性。

圖1
2. 實驗結(jié)果-DNA質(zhì)量評價
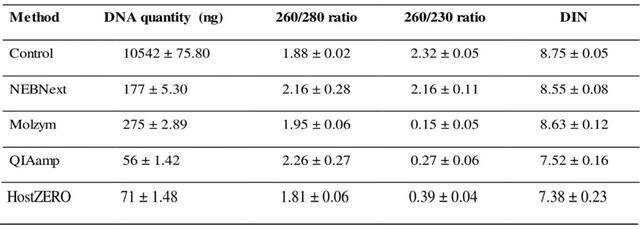
從表1可以看出,五種方法抽提的DNA用NanoDrop™2000c(Thermo Scientific)分光光度法測定提取的DNA產(chǎn)量和純度約為1.8(260/280),基本符合要求。TapeStation 2200顯示DNA完整性(DIN)在7到9之間,這表明DNA沒有碎片化(表1)。而Molzym、QIAamp和HostZERO的260/230比值較低。根據(jù)Qiagen應(yīng)用說明,260/230比率低很可能是由于在基于柱的試劑盒中裂解緩沖液經(jīng)常使用的胍殘留所導(dǎo)致,但這不影響下游qPCR檢測。
表1 不同方法抽提DNA的各項指標(biāo)
3. 實驗結(jié)果-DNA成分鑒定
使用細菌16S特異引物(5’-CCTACGGGAGGCAGCAG-3’和5’-ATTACCGCGGCTGCTGG-3’)和人18S特異引物(5’-GGTGGTGCCCTTCCGTCA-3’ 和 5’-CGATGCGGCGGCGTTATT-3’),通過定量PCR檢測DNA樣本中宿主和細菌DNA的占比,結(jié)果顯示,NEBNext法的宿主DNA污染程度高,HostZERO法和QIAamp法的宿主DNA污染程度低(NEBNext>Molzym>QIAamp>HostZERO)(圖2)對照樣品18S/16S rRNA比值為0.865±0.020。而NEBNext法和Molzym法提取的DNA樣本的18S/16S rRNA比值分別為0.701±0.022和0.393±0.057,仍然比較高。
相比之下,QIAamp法和HostZERO法的18S/16S rRNA比值較低,分別為0.027±0.005和0.015±0.007,分別比對照法低了32倍和57倍。
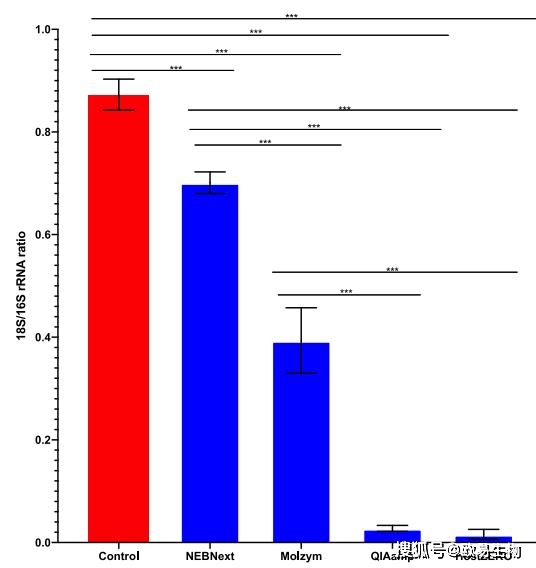
圖2
4. 實驗結(jié)果-微生物DNA占比
通過對定量PCR的數(shù)據(jù)進行拷貝數(shù)計算結(jié)果表明,對照樣品中細菌DNA含量為(6.7±0.1%)。在去除宿主DNA后,NEBNext法的細菌DNA占比仍然比較低(8.1±0.2%),Molzym法略高(13.6±1.0%)。在QiaAmp法(71.0±2.7%)和HostZero法(79.9±3.1%)中,細菌DNA含量增加了10倍以上(圖3)。上述結(jié)果表明,HostZERO法和Qiamp法能有效地去除宿主DNA污染,富集微生物DNA。
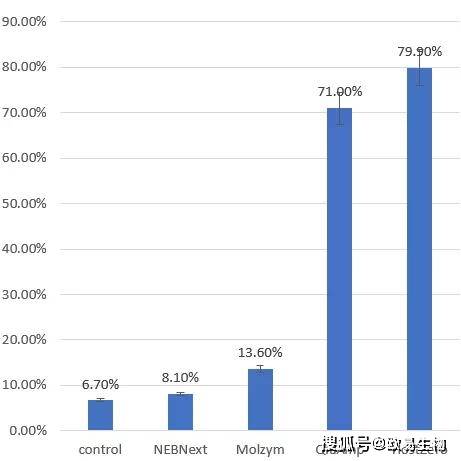
圖3
5. 實驗結(jié)果-16S rRNA擴增測序結(jié)果
使用16S rRNA V3V4區(qū)引物341(5‘-CCTAYGGGRBGCASCAG-3’)和806(3‘-GGACTACNNGGGTATCTAAT-5’)對樣本進行擴增及微生物多樣性檢測,結(jié)果表明:糖尿病足感染組織樣本微生物群共包含5個門,這些門在該樣本中常見。其中,硬壁門和放線桿菌的成員控制著細菌種群(圖4)。NEBNext與對照樣品的細菌物種組成(所有五種報告的菌門的提取)相似,這可能是因為這兩組樣本的DNA提取方法(Roche)一致,NEBNext另外增加了去宿主DNA的步驟。聚類分析也支持這一點,而HostZERO、Molzym和QIAamp形成了一個單獨的聚類(圖4A和圖4B)。在屬一級,鏈球菌主導(dǎo)著糖尿病足感染組織的微生物群,其次是消化鏈球菌(圖4B),與已有報道結(jié)果一致。總的來說,本研究中使用的所有微生物組DNA富集方法都成功地識別了糖尿病足感染樣本中常見的以及臨床上分離出的重要的屬,其豐度與先前研究中的報告相對相似。
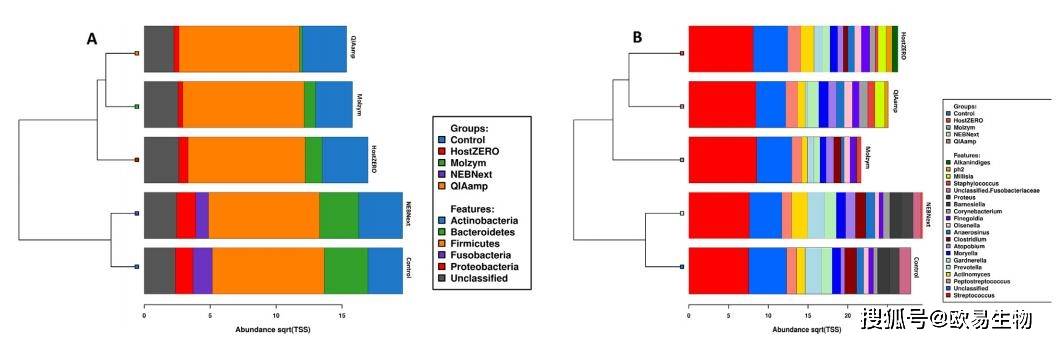
圖4
結(jié)論
由以上研究結(jié)果可知,在測試的五種抽提方法獲得的DNA質(zhì)量相近,但是量的差異比較大。不同的去宿主DNA的方法效果不一,以HostZERO的去宿主效果好,QIAamp的效果其次。用這幾種方法抽提的DNA進行16S rDNA擴增對微生物進行檢測,結(jié)果一致性較高,但是如果用于宏基因組測序,HostZERO和QIAamp抽提方法可能能夠更好的去除宿主基因組的影響。
參考文獻
Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples, Fatemah Sadeghpour Heravi, Martha Zakrzewski, Karen Vickery, et. Al., 2020, Journal of Microbiological Methods, https://doi.org/10.1016/j.mimet.2020.105856
附表
表2 幾種宿主DNA去除方法的優(yōu)劣勢比較

本文轉(zhuǎn)自歐易生物


